
Abstract
Frequent and widespread testing of members of the population who are asymptomatic for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is essential for the mitigation of the transmission of the virus. Despite the recent increases in testing capacity, tests based on quantitative polymerase chain reaction (qPCR) assays cannot be easily deployed at the scale required for population-wide screening. Here, we show that next-generation sequencing of pooled samples tagged with sample-specific molecular barcodes enables the testing of thousands of nasal or saliva samples for SARS-CoV-2 RNA in a single run without the need for RNA extraction. The assay, which we named SwabSeq, incorporates a synthetic RNA standard that facilitates end-point quantification and the calling of true negatives, and that reduces the requirements for automation, purification and sample-to-sample normalization. We used SwabSeq to perform 80,000 tests, with an analytical sensitivity and specificity comparable to or better than traditional qPCR tests, in less than two months with turnaround times of less than 24 h. SwabSeq could be rapidly adapted for the detection of other pathogens.
Similar content being viewed by others


Main
Public health strategies remain essential tools for controlling the spread of SARS-CoV-2, the cause of coronavirus disease 2019 (COVID-19). In contrast to SARS-CoV-1, for which infectivity is associated with symptoms1,2,3,4, the infectivity of SARS-CoV-2 is high during the asymptomatic/presymptomatic phase5,6. As a consequence, containing transmission solely on the basis of symptoms is impossible, which makes molecular screening for SARS-CoV-2 essential for pandemic control.
During the surges of infection in the US, the rise in cases has frequently overwhelmed the capacity of qPCR with reverse transcription (RT–qPCR) tests, which make up the majority of FDA-authorized tests for COVID-19. Delays in obtaining test results, which are due to capacity constraints rather than assay times7, render testing ineffective for the public health aims of preventing viral transmission and suppressing local outbreaks. Even in cases in which expanded testing capacity exists, the ~US$100 price of tests (current Medicare reimbursement rates8) prohibits their widespread adoption by large employers and schools on a regular basis for effective viral suppression9,10. Frequent, low-cost population testing combined with contact tracing and isolation of infected individuals would help to halt the spread of COVID-19 and reopen society11,12. In this Article, we describe SwabSeq—a SARS-CoV-2 testing technology that leverages next-generation sequencing to massively scale up testing capacity13,14.
SwabSeq improves on one-step PCR with reverse transcription (RT–PCR) approaches in several key areas. Similar to other sequencing approaches, SwabSeq uses molecular barcodes that are embedded in the RT–PCR primers to uniquely label each sample and enable the simultaneous sequencing of hundreds to thousands of samples in a single run (LampSeq15, Illumina CovidSeq16, DxSeq17). SwabSeq uses very short reads, reducing sequencing times such that results can be returned in less than 24 h.
To deliver robust and reliable results at scale, SwabSeq adds to every sample a synthetic in vitro RNA standard with a sequence that is nearly identical to the target in the virus genome, but is easily distinguished by sequencing. SARS-CoV-2 detection is based on the ratio of the counts of true viral sequencing reads to those from the in vitro viral standard. As every sample contains the synthetic RNA, SwabSeq controls for the failure of amplification—samples with no SARS-CoV-2 detected are those in which only in vitro viral standard reads are observed, while those without viral or in vitro viral standard reads are inconclusive.
The RNA control confers a number of additional advantages to the SwabSeq assay. As we are interested in only the ratio of real virus to in vitro standard, the PCR can be run to the end point at which all primers are consumed, rather than for a set number of cycles. By driving the reaction to the end point, we overcome the presence of varying amounts of RT and PCR inhibitors and effectively force each sample to have similar amounts of final product. Using in vitro standard RNA with end-point PCR has two important consequences. First, we can pool reaction products after PCR because the yield of product per sample is mostly determined by the primer concentration and not by the sample concentration. Second, we can directly process extraction-free samples. Inhibitors of RT and PCR present in mucosal tissue or saliva should affect both the virus and the in vitro standard equally. End-point PCR overcomes the effect of inhibition, while keeping the ratio of reads between the two RNA species approximately constant, and therefore avoids the need for extraction.
Here we show that SwabSeq has extremely high sensitivity and specificity for the detection of viral RNA in purified samples. We also demonstrate a low limit of detection in extraction-free lysates from mid-nasal swabs and saliva. Finally, we describe our real-world use of SwabSeq to perform more than 80,000 tests for COVID-19 in less than two months in a high-complexity Clinical Laboratory Improvement Amendments (CLIA) certified laboratory. These results demonstrate the potential of SwabSeq to be used for SARS-CoV-2 testing on an unprecedented scale, offering a potential solution to the need for population-wide testing to stem the pandemic.
Results
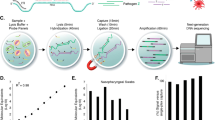
SwabSeq is a simple and scalable protocol that consists of five steps (Fig. 1a): (1) sample collection; (2) reverse transcription and PCR using primers that contain unique molecular indices at the i7 and i5 positions (Fig. 1b and Supplementary Fig. 1) as well as in vitro standards; (3) simple pooling (no normalization) and clean-up of the uniquely barcoded samples for library preparation; (4) sequencing of the pooled library; and (5) computational assignment of barcoded sequencing reads to each sample for counting and viral detection.

a, The workflow for SwabSeq is a five-step process that takes approximately 12 h from start to finish. b, In each well, we perform RT–PCR on clinical samples. Each well has two sets of indexed primers that generate cDNA and amplicons for the SARS-CoV-2 S gene and the human RPP30 gene. Each primer is synthesized with the P5 and P7 adaptors for Illumina sequencing, unique i7 and i5 molecular barcodes, and the unique primer pair. Importantly, every well has a synthetic in vitro S standard that is key to enabling the method to work at scale. c, The in vitro S standard differs from the virus S gene by 6 bp that are complementary (underlined). d, Read count at various viral concentrations. e, Ratiometric normalization enables in-well normalization for each amplicon. f, Every well has two internal well controls for amplification, the in vitro S standard and the human RPP30 standard. The RPP30 amplicon serves as a control for specimen collection. The in vitro S standard is critical for the ability of SwabSeq to distinguish true negatives.
Our assay consists of two primer sets that amplify two genes—the S gene of SARS-CoV-2 and the human ribonuclease P/MRP subunit P30 (RPP30). We include a synthetic in vitro RNA standard that is identical to the viral sequence targeted for amplification, except for the most upstream 6 bp (Fig. 1c), which enables us to distinguish between sequencing reads that correspond to the in vitro standard and those that correspond to the target sequence. The primers amplify both the viral and the synthetic sequences with equal efficiency (Supplementary Fig. 2). We also designed a second RNA standard for RPP30 with a similar design. The ratio of the number of native reads to the number of in vitro standard reads provides a more accurate and quantitative measure of the number of viral genomes in the sample than native read counts alone (Fig. 1d,e). The in vitro standard also enables us to retain linearity over a large range of viral input despite the use of end-point PCR (Supplementary Fig. 3). Using this approach, the final amount of DNA in each well is largely defined by the total primer concentration rather than by the viral input—negative samples have higher amounts of in vitro S standard and lower/zero amounts of viral reads, and positive samples have lower amounts of in vitro S standard and higher amounts of viral reads (Supplementary Fig. 3). In addition to viral S, we reverse-transcribe and amplify a human housekeeping gene to control for specimen quality, as in traditional qPCR assays (Fig. 1f). We also assign i5 and i7 sample barcodes that are designed to be at least several base-pair edits away from one another, allowing for demultiplexing even in the case of sequencing errors.
After RT–PCR, samples are combined at equal volumes, purified and used to generate one sequencing library. We use the Illumina MiSeq, MiniSeq and the Illumina NextSeq 550 systems to sequence these libraries (Supplementary Fig. 4). We minimize instrument sequencing time by sequencing only the minimum required 26 bp (Methods). Each read is classified as deriving from native or in vitro standard S or RPP30, and assigned to a sample on the basis of the associated index sequences (barcodes). To maximize the specificity and avoid false-positive signals arising from incorrect classification or assignment, conservative edit-distance thresholds are used for this matching operation (Methods and Supplementary Results). A sequencing read is discarded if it does not match one of the expected sequences. Counts for native and in vitro S standard and RPP30 reads are obtained for each sample and used for downstream analyses (Methods).
We have estimated that approximately 5,000 reads per well are sufficient to detect the presence or absence of viral RNA in a sample (Methods). This translates to at least 1,500 samples per run on a MiSeq or MiniSeq, 20,000 samples per run on a NextSeq 550 and up to 150,000 samples per run on a NovaSeq S2 flow cell. Computational analysis takes only minutes per run18. The MiniSeq newly released high output rapid flow cell has a turnaround time of 2.5 h, which substantially improves our throughput and turnaround time. Our optimized protocols enable a single person to process 6–384-well plates in a single hour, which is equivalent to 2,304 samples per hour. This process cloned over multiple liquid handlers can rapidly scale to 10,000 samples per hour with a staff size of 6 people. In our current operations, we routinely run up to 1,536 samples per flow cell on a MiniSeq.
We have optimized the SwabSeq protocol by identifying and eliminating multiple sources of noise (Supplementary Results) to create a streamlined and scalable protocol for SARS-CoV-2 testing.
Validation of SwabSeq as a diagnostic technology
We first validated SwabSeq on purified RNA nasopharyngeal samples that were previously tested by the UCLA Clinical Microbiology Laboratory using a standard RT–qPCR assay (Taqpath COVID19 Combo Kit). To determine our analytical limit of detection, we diluted inactivated virus with pooled, remnant clinical nasopharyngeal swab specimens. The remnant samples were all confirmed to be negative for SARS-CoV-2. In these remnant samples, we performed a serial twofold dilution of heat-inactivated SARS-CoV-2 (ATCC, VR-1986HK) from 8,000 to 125 genome copy equivalents (GCE) per ml. We detected SARS-CoV-2 in 34/34 samples down to 250 GCE per ml, and in 28/34 samples down to 125 GCE per ml (Fig. 2a). These results established that SwabSeq is highly sensitive, with an analytical limit of detection of 250 GCE per ml for purified RNA from nasal swabs. This limit of detection is lower than the limit of detection of many currently FDA-authorized and highly sensitive RT–qPCR assays for SARS-CoV-2.

a, Limit of detection in nasal swab samples with no SARS-CoV-2 that were pooled and then ATCC inactivated virus was added at different concentrations. Nasal swab samples were RNA-purified and tested using SwabSeq, showing a limit of detection of 250 GCE per ml. b, RNA-purified clinical nasal swab specimens obtained through the UCLA Health Clinical Microbiology Laboratory were tested on the basis of clinical protocols using FDA-authorized systems and then also tested using SwabSeq. This represents a subset of the total purified RNA samples used in our validation. We show 100% agreement with samples that tested positive for SARS-CoV-2 (n = 63) and negative for SARS-CoV-2 (n = 159). c, We also tested RNA-purified samples from extraction-free nasopharyngeal swabs and showed a limit of detection of 558 GCE per ml. d, The relationship between Ct from RT–qPCR targeting the S gene (x axis) and the SwabSeq ratio for extraction-free swabs in normal saline or TE buffer (y axis) for patient samples classified as testing positive or negative for SARS-CoV-2 by the UCLA Clinical Microbiology Laboratory. Samples with no virus detected were assigned a Ct value of 0 for this visualization. e, Extraction-free processing of saliva specimens shows a limit of detection down to 1,000 GCE per ml. f, Extraction-free processing of saliva clinical specimens using SwabSeq (y axis) compared to classification of SARS-CoV-2 status from RNA-purified clinical nasal swab specimens for matched samples (x axis).
SwabSeq detects the SARS-CoV-2 genome with a high clinical sensitivity and specificity. We retested 380 RNA-purified nasopharyngeal samples from the UCLA Clinical Microbiology Laboratory comprising n = 94 SARS-CoV-2-positive samples and n = 286 SARS-CoV-2-negative samples. We observed 99.2% agreement with RT–qPCR results for all of the samples (Fig. 2b and Supplementary Fig. 5d) with 100% concordance in samples that were positive for SARS-CoV-2. We sequenced these samples using a MiSeq, MiniSeq or NextSeq550 system (Supplementary Fig. 5), with 100% concordance between the different sequencing instruments.
One of the major bottlenecks in scaling up RT–qPCR diagnostic tests is the RNA-purification step. RNA extraction is challenging to automate, making it a time-consuming and labour-intensive step, and supply chains have not been able to keep up with the demand for the necessary reagents during the course of the pandemic. Thus, we examined the ability of SwabSeq to detect SARS-CoV-2 directly from a variety of extraction-free sample types. The CDC recommends several types of medium for nasal swab collection: viral transport medium19, Amies transport medium20 and normal saline20. A main technical challenge is RT or PCR inhibition by ingredients in these collection buffers. We found that diluting specimens with water overcame the RT and PCR inhibition and enabled us to detect viral RNA in contrived and positive clinical patient samples, with higher limits of detection of between 4,000 and 6,000 GCE per ml (Supplementary Fig. 6). We also tested nasal swabs that were collected directly into Tris-EDTA (TE) buffer diluted 1:1 with water. This approach yielded a limit of detection of 560 GCE per ml (Fig. 2c). We next performed a comparison between traditional purified RT–qPCR protocols and our extraction-free protocol for nasopharyngeal samples collected into either normal saline (n = 128) or TE (n = 170). We showed 96.0% overall agreement, 92.0% positive percent agreement and 97.6% negative percent agreement for all samples and a high correlation (r2 = 0.68) between SwabSeq signal and RT–qPCR, and high reproducibility at the lower end of our limit of detection (Fig. 2d and Supplementary Figs. 7–9).
We also tested extraction-free saliva protocols in which saliva is collected directly into a matrix tube using a funnel-like collection device (Fig. 1a). The main technical challenges in demonstrating the detection of virus in saliva samples have been preventing the degradation of the inactivated SARS-CoV-2 virus that is added to saliva and ensuring accurate pipetting of this heterogeneous and viscous sample type. We found that heating the saliva samples to 95 °C for 30 min (ref. 21) reduced PCR inhibition and improved the detection of the S amplicon (Supplementary Fig. 10). After heating, we diluted samples at a 1:1 volume with 2× TBE with 1% Tween-20 (ref. 21). Using this method, we obtained a limit of detection of 1,000 GCE per ml (Fig. 2e). In a study performed in the UCLA Emergency Department, we collected paired saliva and nasopharyngeal swab samples in patients with COVID-19-like symptoms and compared our extraction-free, saliva SwabSeq protocol and purified nasopharyngeal swab samples run with high-sensitivity RT–qPCR in the UCLA Clinical Microbiology laboratory. We showed 95.3% overall agreement, 90.2% positive percent agreement and 96.3% negative percent agreement across 537 samples. (Supplementary Fig. 11).
Deployment of SwabSeq for large-scale asymptomatic screening
After obtaining a CLIA license on 30 October 2020, we carried out a pilot phase of clinical testing starting in November 2020, during which we tested 8,199 samples over 6 weeks (Fig. 3a). Following the success of this pilot phase, we began large-scale testing at the start of 2021 to support asymptomatic screening of health care workers at a major academic medical center as well as students and staff at local colleges and universities. To date, we have returned results for over 80,000 tests, at a scale of ~10,000 tests per week, with an overall asymptomatic positivity rate of 0.67% (the weekly positivity rate during this time period ranged from 0.3% to 2.9%). Our automated high-throughput protocol has enabled us to achieve this scale with a lightly staffed laboratory of around six personnel. Here, we describe the innovations and optimizations that enable us to test tens of thousands of samples per week (Fig. 3).

a, Since November 2020, we have used SwabSeq for large-scale screening in conjunction with our saliva and nasal swab collection processes. We have scaled up to nearly 10,000 samples per week and are continuing to increase our capacity. Week number refers to the week of the year spanning 2020–2021. Our weekly percentage positivity rate ranges between 0.3% and 2.4% over this period. b, Our clinical deployment streamlined the pre-analytical testing process such that we receive tubes that are ready to be processed through our testing protocol.
Sample registration is one of the most labour-intensive aspects of obtaining samples. We partnered with PreciseMDX and used their website for online ordering of tests and return of results (Supplementary Fig. 12). We tested both self-collected nasal swab and saliva samples (Fig. 3b); collection was coordinated by the testing site using kits provided by our laboratory. A sample is registered to an order when barcodes on the sample tubes are scanned using the PreciseMDX software at the time of collection. In doing so, we eliminated the need for laborious and error-prone manual pipetting of samples into uniform containers for testing.
We further improved the efficiency of testing at scale by requiring participants to use one of two sample collection kits that are compatible with downstream automation. In one kit, saliva samples are collected by custom funnels into tubes that are then racked in a 96-tube format at the collection site and can be uncapped using automated decappers. This enables us to pipet batches of 96 samples at a time directly into a RT–PCR mix (Fig. 3b). Our second collection kit uses a nasal swab that we designed that has a break-off point just below the tip. The sample can therefore be processed with the swab without interfering with our downstream testing processes (Supplementary Fig. 13).
Translation of SwabSeq from the bench to real-world testing required process optimization. The development of extraction-free protocols prioritized ‘low-touch’ handling. Our inactivation protocol requires simple heating of the specimen tube, minimizing exposure and pipetting of the sample. We limited our use of plastic consumables, and used only one liquid-handler tip per sample, thereby minimizing supply chain challenges. Finally, we moved to single-use, index primer plates to eliminate the variability in primer pipetting when this is performed manually.
How many samples can be processed? We calculate that a single person can process six 384-well plates per hour using an automated liquid handler. In our COVID-19 testing laboratory, we scaled this to four liquid handlers capable of processing up to 24 plates per hour, which is the equivalent to a capacity of 9,216 samples per hour. A laboratory running 24 h per day could handle more than 100,000 samples per day. Furthermore, the post-PCR multiplexing and library generation is designed such that a single person can handle thousands of samples concurrently; the number of samples is dependent only on the capacity of the sequencer. For smaller-scale university settings, testing of up to ~3,000 samples a day would require an automated decapper, a single liquid handler and a MiniSeq, an investment of less than $150,000 in equipment. We provide an R package for fully automated sequence analysis, quality-control reporting and result interpretation.
These optimizations enable rapid scaling of the SwabSeq diagnostic technology at the onset of a novel pandemic. We tested sample collection and processing workflows in a variety of settings, including the emergency room, return-to-school testing and university-wide population screening. These and other optimizations demonstrate a path to rapid scaling in population-dense settings such as university campuses.
Discussion
SwabSeq may alleviate existing bottlenecks in diagnostic clinical testing for Covid-19. We believe that it has an even greater potential to enable testing on a scale necessary for pandemic suppression through population surveillance. The technology uses massively parallel next-generation sequencing for infectious-disease surveillance and diagnostics. We have demonstrated that SwabSeq can detect SARS-CoV-2 RNA in clinical specimens from both purified RNA and extraction-free lysates, with clinical and analytical sensitivity and specificity that is comparable to RT–qPCR performed in a clinical diagnostic laboratory. We have optimized SwabSeq to prioritize scale and low cost, as these are the key factors that are missing from current COVID-19 diagnostics.
Methods for surveillance testing, such as SwabSeq, should be evaluated differently to those used for clinical testing. Clinical testing informs medical decision-making, and therefore requires high sensitivity and specificity. For surveillance testing, the important factors are the breadth and frequency of testing and the turn-around-time11. Sufficiently broad and frequent testing with a rapid return of results, contact tracing and quarantining of infectious individuals can effectively contain viral outbreaks, avoiding blanket stay-at-home orders. Epidemiological modelling of surveillance testing on university campuses has shown that diagnostic tests with only 70% sensitivity, performed frequently with a short turn-around time, can suppress transmission12. However, there remain major challenges for practical implementation of frequent testing, including the cost of testing and the logistics of collecting and processing thousands of samples per day.
The use of next-generation sequencing in diagnostic testing has garnered concern about turn-around-time and cost. SwabSeq uses short sequencing runs that read out the molecular indexes and 26 bp of the target sequence in as little as 3 h, followed by computational analysis that can be performed on a desktop computer in 5 min. The cost of 1,000 samples analysed in one MiSeq run is less than US$1 per sample for sequencing reagents. Running 10,000 samples on a NextSeq550, which generates 13 times more reads per flow cell, can reduce this sequencing cost by approximately tenfold. We estimate that the total consumable cost, inclusive of the collection kit and informatics infrastructure, ranges from US$4 to US$6 per test. The total cost of operations depends on a wide range of factors, including the costs of setting up a CLIA-certified laboratory, certified personnel, logistics of sample collection and result reporting. Ongoing optimization to decrease reaction volumes and use less-expensive RT–PCR reagents can further decrease the total cost per test.
Finally, scaling up testing for SARS-CoV-2 requires high-throughput sample collection and processing workflows. Manual processes, which are common in most academic clinical laboratories, are not easily compatible with simple automation. The current protocols with nasopharyngeal swabs into viral transport medium, Amies buffer or normal saline are collection methods that date back to the pre-molecular-genetics era, when live viral culture was used to identify cytopathic effects on cell lines. A fresh perspective on collection methods that are easily scalable is beneficial to scaling up centralized laboratory testing approaches.
We have successfully demonstrated that it is possible to set up SwabSeq in a small testing laboratory to process tens of thousands of samples per week, and with appropriate instrumentation to scale to hundreds of thousands. To promote scalability, we have developed sample-collection protocols that use smaller-volume tubes that are compatible with simple automation, such as automated capper–decappers and 96-head liquid handlers22,23. These approaches decrease the amount of hands-on work required in the laboratory to process tests and enable a higher reproducibility, faster turnaround time and decreasing exposure risk to laboratory workers. However, most of the challenges to scaling lie in developing interfaces for efficient sample registration, collection of patient information and return of results. We have shown one way to overcome these challenges, but each laboratory will face unique challenges due to the need to meet whatever regulatory requirements are in place, and the need to integrate with existing systems for handling patient information.
Outlook
The SwabSeq technology complements traditional clinical diagnostics tests24, as well as the growing arsenal of point-of-care rapid diagnostics25 emerging for COVID-19, by increasing test capacity to meet the needs of both diagnostic and widespread surveillance testing. SwabSeq is easily extensible to accommodate additional pathogens and viral targets. This would be particularly useful during cold and flu seasons, when multiple respiratory pathogens circulate in the population and cannot be easily differentiated on the basis of symptoms alone. Furthermore, to track the increasing number of SARS-CoV-2 variant strains, we can sequence additional viral amplicons to genotype key mutations and to further improve test sensitivity (Supplementary Fig. 22). We believe that SwabSeq could be rapidly established within several weeks to accommodate future novel pathogens with pandemic potential. Surveillance testing is likely to become a part of a new normal as the educational, business and recreational sectors of society are safely reopened.
Methods
Sample collection
All patient samples used in our study were deidentified. All samples were obtained with UCLA IRB approval. Nasopharyngeal samples were collected by healthcare providers from individuals who physicians suspected had COVID-19. After deployment of testing, we obtained clinical specimens and tested them under our CLIA license (05D2198285) with a laboratory developed test.
Creation of contrived specimens
For the clinical limit-of-detection experiments, we pooled confirmed, COVID-19 negative remnant nasopharyngeal swab specimens collected by the UCLA Clinical Microbiology Laboratory. Pooled clinical samples were then spiked with ATCC Inactivated Virus (ATCC, 1986-HK) at specified concentrations and extracted as described below. For the clinical purified RNA samples, they were collected as nasopharyngeal swabs and purified using the KingFisherFlex (Thermo Fisher Scientific) instrument using MagMax bead extraction. All extractions were performed according to the manufacturer’s protocols. For extraction-free samples, we first contrived samples at specified concentrations into pooled, confirmed negative clinical samples and diluted samples in TE buffer or water before adding to the RT–PCR master mix.
Processing of extraction-free saliva specimens
Direct saliva was collected into a Matrix tube (Thermo Fisher Scientific, 3741-BR) using a small funnel (TWDRer, 6565). The saliva samples were collected into a matrix tube and heated to 95 °C for 30 min. Samples were then either frozen at −80 °C or processed by dilution with 2× TBE with 1% Tween-20, for a final concentration of 1× TBE and 0.5% Tween-20 (ref. 21). We also tested 1× Tween-20 with Qiagen Protease and RNA Secure (Thermo Fisher Scientific), which also works but resulted in more sample-to-sample variability and required additional incubation steps.
Processing of extraction-free nasal swab lysates
All extraction-free lysates were inactivated using heat inactivation at 56 °C for 30 min. Samples were then diluted with water at a ratio of 1:4 and directly added to the master mix. Dilution amounts varied depending on the liquid medium that was used. We found that, of the CDC recommended media, normal saline performed the most robustly. Viral transport media and Amies buffer showed substantial PCR inhibition that was difficult to overcome, even with dilution in water. We recommend placing the swab directly into the diluted TE buffer, which has little PCR inhibition.
Barcode primer design
Barcode primers were chosen from a set of 1,536 unique 10 bp i5 barcodes and a set of 1,536 unique 10 bp i7 barcodes. These 10 bp barcodes satisfied the criteria that there is a minimum Levenshtein26 distance of 3 between any two indices (within the i5 and i7 sets) and that the barcodes contain no homopolymer repeats of greater than 2 nucleotides. Furthermore, barcodes were chosen to minimize homo- and heterodimerization using helper functions in the python API to Primer3 (ref. 27).
The specificity of each of our primer pairs was tested against 38 other common respiratory viruses, including MERS and SARS-CoV-1 using BLASTn, and no notable homology (defined as >80% homology) was identified. Additional details and code for primer design and specificity analysis are available at GitHub (https://github.com/octantbio/SwabSeq).
Construction of the S and RPP30 in vitro standards
RT–PCR was performed using the primers shown in Supplementary Table 1 on gRNA of SARS-CoV-2 (Twist BioSciences, 1) for construction of a in vitro S standard DNA template. RT–PCR (FP_1, R) and a second round of PCR (FP_2, R) was performed on HEK293T lysate to construct an in vitro RPP30 standard DNA template. Products were run on a gel to identify specific products at ~150 bp. DNA was purified using Ampure beads (Axygen) using a 1.8 ratio of beads:sample volume. The mixture was vortexed and incubated for 5 min at room temperature. A magnet was used to bind beads for 1 min, washed twice with 70% ethanol, beads were air-dried for 5 min, and then removed from the magnet and eluted in 100 μl of IDTE Buffer. The bead solution was placed back onto the magnet and the eluate was removed after 1 min. DNA was quantified using the Nanodrop (Denovix) system.
This prepared DNA template was used for standard HiScribe T7 in vitro transcription (NEB). In vitro transcription reactions prepared according to the manufacturer’s instructions using 300 ng of template DNA per 20 μl reaction with a 16 h incubation at 37 °C. In vitro transcription reactions were treated with DNase I according to the manufacturer’s instructions. RNA was purified using the RNA Clean & Concentrator-25 kit (Zymo Research) according to the manufacturer’s instructions and eluted into water. The RNA standard was quantified both using the Nanodrop and the RNA screen tape kit for the TapeStation according to the manufacturer’s instructions (Agilent) to verify that the RNA was the correct size (~133 nucleotides).
Construction of the S-diversified synthetic spike-in standard
The diversified S standard is a control for reverse transcription and amplification of the viral sequence in each RT–PCR well. The diversified S standard was designed to have a similar sequence to the S amplicon, but each spike has a unique 26 bp region that is read by the sequencer that enables improved base-calling and removes the reliance on PhiX loading. This was achieved by ensuring equal base coverage at each position along the 26 bp region. This amplicon can be differentiated from the true viral S amplicon by sequencing. The S primer pair will amplify both the S amplicon and the S-diversified standard, at an equivalent efficiency. These RNA oligonucleotides were custom synthesized by Synthego and were supplied as four separate tubes of equal concentration, and were mixed in equal ratios before testing. This equimolar mix of RNA was spiked-in every well before RT–PCR.
One-step RT–PCR
RT–PCR reactions were performed using either the Luna Universal One-Step RT-qPCR Kit (New England BioSciences, E3005) or the TaqPath 1-Step RT-qPCR Master Mix (Thermo Fisher Scientific, A15300) with a reaction volume of 20 μl. Both kits were used according to the manufacturer’s protocol. The final concentration of primers in our master mix was 50 nM for the RPP30 F and R primers and 400 nM for the S F and R primers. Synthetic S RNA was added directly to the master mix at a copy number of 250–500 copies per reaction. We recommend 250 copies per reaction for extraction-free samples and 500 copies for extracted samples. The sample was loaded into a 20 μl reaction. All reactions were run in a 96- or 384-well format and thermocycler conditions were run according to the manufacturer’s protocol. We observed substantial differences between the amplification of samples from purified RNA versus extraction-free (unpurified swab) samples (Supplementary Fig. 14). For purified RNA samples, we performed 40 cycles of PCR. For extraction-free samples, we performed end-point PCR for 50 cycles.
Multiplex library preparation
After the RT–PCR reaction, samples were pooled using a multichannel pipet or Integra Viaflow Benchtop liquid handler. Then, 6 μl from each well was combined in a sterile reservoir and transferred into a 15 ml conical tube and vortexed. Next, 100 μl of the pool was transferred into a 1.7 ml Eppendorf tube for a double-sided solid phase reversible immobilization clean-up28. In brief, 50 μl of AmpureXP beads (Beckman Coulter, A63880) was added to 100 μl of the pooled PCR volume and vortexed. After 5 min, a magnet was used to collect beads for 1 min and supernatant transferred to a new Eppendorf tube. An additional 130 μl of Ampure XP beads was added to the 150 μl of supernatant and vortexed. After an additional 5 min, the magnet was used to collect beads for 1 min and the beads were washed twice with fresh 70% ethanol. DNA was eluted off the beads in 40 μl of Qiagen EB buffer. The magnet was used to collect beads for 1 min and 33 μl of supernatant was transferred to a new tube. Purified DNA was quantified and the library quality was assessed using the Agilent TapeStation. We observed some differences in non-specific peaks in our TapeStation analysis of the final library preparation, particularly when sequencing extraction-free samples out to 50 cycles (Supplementary Fig. 15). The presence of non-specific products affects the quantification of the library, loading concentration and cluster density.
Sequencing protocol with the original S standard
Libraries were sequenced using the Illumina MiSeq (2012), NextSeq550 or MiniSeq system. Before each MiSeq run, a bleach wash was performed using a sodium hypochlorite solution (Sigma Aldrich, 239305) according to Illumina protocols. We also performed a maintenance wash between each run. On the NextSeq550 and MiniSeq systems, the post-run wash was performed automatically by the instruments, and no human intervention was required. The pooled and quantified library was diluted to a concentration of 6 nM (based on Qubit 4 Fluorometer and Illumina’s formula for conversion between ng μl−1 and nM) and was loaded on the sequencer at either 25 pM (MiSeq), 1.35 pM (NextSeq) or 1.6 pM (MiniSeq). PhiX Control v3 (Illumina, FC-110-3001) was spiked into the library at an estimated 30–40% of the library. PhiX provides additional sequence diversity to read 1, which assists with template registration and improves run and base quality.
For this application, the MiSeq and MiniSeq (Rapid Run Kit) systems require two custom sequencing primer mixes, the read 1 primer mix and the i7 primer mix. Both mixes have a final concentration of 20 μM of primers (10 μM of each amplicon’s sequencing primer). The NextSeq system requires an additional sequencing primer mix, the i5 primer mix, which also has a final concentration of 20 μM. The MiSeq Reagent Kit v3 (150-cycle; MS-102-3001) was loaded with 30 μl of read 1 sequencing primer mix into reservoir 12 and 30 μl of the i7 sequencing primer primer mix into reservoir 13. The Miniseq Rapid High Output Reagent Kit (100-cycle; 20044338) was loaded with 17 μl of read 1 sequencing primer mix into reservoir 24 and 26 μl of i7 sequencing primer mix into reservoir 28. The NextSeq 500/550 Mid Output Kit (150 cycles; 20024904) was loaded with 52 μl of read 1 sequencing primer mix into reservoir 20, 85 μl of i7 sequencing primer mix into reservoir 22 and 85 μl of i5 sequencing primer mix into reservoir 22. Index 1 and 2 are each 10 bp, and read 1 is 26 bp.
Sequencing protocol with the optimized-diversity S standard
The pooled and quantified library was diluted to a concentration of 5 nM (using the High Sensitivity DNA Qubit and Illumina’s formula for conversion between ng μl−1 and nM assuming 195 bp for the length of the library). Without PhiX, we observed more consistent sequencing results if we underloaded the sequencers to 83% of the recommended loading concentrations specified by Illumina. Libraries were loaded onto the sequencer at either 1.25 pM (NextSeq) or 1.5 pM (MiniSeq). Custom sequencing primer mixes were prepared and loaded onto sequencers as described in the section above, except we loaded a larger volume of the read 1 primer mix (24.5 μl) onto the MiniSeq.
Diversified N1 spike and primer design
The N1 amplicon was designed using the CDC RT–PCR primer sequences for SARS-CoV-2 (2019-nCoV_N1-F, GACCCCAAAATCAGCGAAAT; and 2019-nCoV_N1-R; TCTGGTTACTGCCAGTTGAATCTG). Diversified N1 spikes were designed using the same principles as the S diversified spike design: four spike sequences with a conserved homology arm in the primer and Illumina read 1 oligonucleotide-binding region but diversified 26 bp read region ensuring equal base coverage in each sequencing cycle.
FluA and FluB primer design
To implement SwabSeq for other respiratory pathogens, we designed universal influenza A and influenza B SwabSeq primers. For influenza A, we designed and tested 2 primer pairs targeting influenza A M1 (FluA_5_f1, GACCAATCYTGTCACCTCTGAC; FluA_5_f2, GACCAATYCTGTCACCTYTGAC; FluA_5_r, AGGGCATTTTGGAYAAAGCGTCTA) and NS1 (FluA_12_f, TTGGGGTCCTCATCGGAG; FluA_12_r, TTCTCCAAGCGAATCTCTGTA). For influenza B, we designed and tested a primer pair specific for NS1 (FluB_11_f, AAGATGGCCATCGGATCC; FluB_11_r, GTCTCCCTCTTCTGGTGATAATC). An in silico cross-reactivity analysis was performed as described for the S and RPP30 primers, none of the pathogens exhibit greater than 80% homology with any of the primers.
Bioinformatics analysis
The bioinformatics analysis consists of the standard conversion of BCL files into FASTQ sequencing files using Illumina’s bcl2fastq (v.2.20.0.422). Demultiplexing and read counts per sample was performed using our custom software. Here read 1 is matched to one of the three expected amplicons enabling the possibility of a single-nucleotide error in the amplicon sequence. The Hamming distance is the number of positions at which the corresponding sequences are different from each other and is a commonly used measure of the distance between sequences. Samples were demultiplexed using the two index reads to identify which sample the read originated from. Observed index reads were matched to the expected index sequences enabling the possibility of a single-nucleotide error in one or both of the index sequences. The set of three reads was discarded if both index 1 and index 2 have Hamming distances of greater than 1 from the expected index sequences. The count of reads for each amplicon and each sample was calculated. In this analysis, we make use of a few custom scripts written in R that rely on the ShortRead29 and stringdist30 packages for processing FASTQ files and calculating the Hamming distances between observed and expected amplicons and indices. This approach was conservative and gave us very low level control of the sequencing analysis. However, we anticipate that continued development of the kallisto and bustools SwabSeq analysis tools31 will provide a more user-friendly and computationally efficient solution for SwabSeq.
We developed an R package (swabseqr) to fully automate sequence analysis, quality-control reporting and result interpretation.
Criteria for the classification of RNA-purified patient samples
For our analytic pipeline, we developed quality control metrics for each type of specimen. For purified RNA, we require each sample to have at least 10 reads detected for RPP30 and that the sum of S and in vitro S standard reads exceeds 2,000 reads. If these conditions are not met, the sample is rerun one time and, if there is a second fail, we request a resample. To determine whether SARS-CoV-2 is present, we calculate whether the ratio of S to in vitro S standard exceeds 0.003 (note that we add 1 count to both the S and in vitro S standard before calculating this ratio to facilitate plotting the results on a logarithmic scale). If the ratio is greater than 0.003, we conclude that SARS-CoV-2 is detected for that sample and, if it is less than or equal to 0.003, we conclude that SARS-CoV-2 is not detected (Supplementary Fig. 5c).
The same pair of primers will amplify both the S and in vitro S standard amplicons. As we run an endpoint assay, the primers will be the limiting reagent to continued amplification. In developing this assay, we observed that, as S counts increase for a sample, the in vitro S standard counts decrease (Supplementary Fig. 3). We found that, at very high viral levels, in vitro S standard read counts decreased to less than 1,000 reads. Thus, analysis of S and in vitro S standard together enabled our quality control to call SARS-CoV-2 even at extremely high viral levels.
As the S and in vitro S standard are derived from the same primer pair, to account for the scenario in which the in vitro S standard counts are low because S amplicon counts are very high and the sample contains large amounts of SARS-CoV-2 RNA (Supplementary Fig. 3), in the QC, we require that the sum of S and in vitro S standard counts together exceeds 2,000. For example, if we detected greater than 2,000 S counts and 0 in vitro S standard counts this would certainly be a SARS-CoV-2-positive sample and we would report the result: SARS-CoV-2 detected.
Criteria for the classification of extraction-free patient samples
We require that the sum of S and S synthetic spike-in reads exceeds 500 reads, or the results are considered to be inconclusive. With inhibitory lysates, we have observed that this lower total is acceptable for maintaining sensitivity and specificity while limiting the number of tests that are considered to be inconclusive. If the sum of S and S synthetic spike-in reads exceeds 500, we determine whether SARS-CoV-2 is detected in a sample by examining whether the ratio of S to S standard exceeds 0.05 (note that we add 1 count to both S and S standard before calculating this ratio to facilitate plotting the results on a logarithmic scale). If the ratio is greater than 0.05, we conclude that SARS-CoV-2 is detected for that sample and, if it is less than or equal to 0.05, we conclude that SARS-CoV-2 is not detected provided that 10 reads are detected for RPP30 for that sample. This serves as a control that sample collection took place properly and contains a human specimen. If fewer than 10 reads are detected for RPP30 and the ratio of S to S standard is less than or equal to 0.05, the results are considered to be inconclusive. We have modified these criteria such that samples without a SARS-CoV-2 signal are considered to be inconclusive only if RPP30 reads are fewer than 10. This ensures that we do not miss SARS-CoV-2-positive samples that may have fewer RPP30 reads (Supplementary Fig. 8).
Downsampling analysis
Reads were downsampled from the results for the nasopharyngeal purified confirmatory limit of detection shown in Supplementary Fig. 5b. We observed that downsampling down to 5,000 reads per well resulted in no instances of misclassification of SARS-CoV-2 presence or absence. At 5,000 reads per well, approximately 3% of wells would no longer pass the filter that the sum of S and S standard reads exceeded 1,000 reads and would result in a sample being classified as inconclusive. A logistic regression classifier described elsewhere18 should robustly tolerate a small fraction of outlier samples with slightly lower read depth.
Analysis of index misassignment
Unique dual indices and amplicon-specific indices were used to study index misassignment. In this scheme, each sample was assigned two unique indices for the S or S standard amplicon and two unique indices for the RPP30 amplicon for a total of four unique indices per sample. A count matrix with all possible pairwise combinations, that is, a ‘matching matrix’, was generated for each index pair (one i7 and one i5) using kallisto and bustools32. The counts on the diagonal of the matching matrix correspond to input samples and counts off the diagonal correspond to index swapping events. The extent of index misassignment for the i7 and i5 index was determined by computing the row and column sums, respectively, of the off-diagonal elements of the matching matrix. The observed rate of index swapping to wells with a known zero amount of viral RNA was determined by computing the mean of the viral S counts to S standard ratio for those wells.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. Source data for all figures are available at GitHub (https://github.com/joshsbloom/swabseq). All protocols and primers are available under an Open COVID License online (https://www.notion.so/Octant-COVID-License-816b04b442674433a2a58bff2d8288df). Videos of the workflow for SwabSeq assay are available at Figshare (https://figshare.com/projects/Additional_SwabSeq_Data/113643).
Code availability
All code can be accessed at GitHub (https://github.com/joshsbloom/swabseq). An R package to automate the diagnosis of patient samples is available at GitHub (https://github.com/joshsbloom/swabseqr). Codes for primer design and for the analysis of cross-reactivity can be found at GitHub (https://github.com/octantbio/SwabSeq). The core technology has been made available under the Open COVID Pledge, and software and data under the MIT license (UCLA) and Apache 2.0 license (Octant).
References
Furukawa, N. W., Brooks, J. T. & Sobel, J. Evidence supporting transmission of severe acute respiratory syndrome coronavirus 2 while presymptomatic or asymptomatic. Emerg. Infect. Dis. 26, e1–e6 (2020).
Lavezzo, E. et al. Suppression of a SARS-CoV-2 outbreak in the Italian municipality of Vo'. Nature 584, 425–429 (2020).
Peiris, J. S. M., Yuen, K. Y., Osterhaus, A. D. M. E. & Stöhr, K. The severe acute respiratory syndrome. N. Engl. J. Med. 349, 2431–2441 (2003).
Cheng, C., Wong, W.-M. & Tsang, K. W. Perception of benefits and costs during SARS outbreak: an 18-month prospective study. J. Consult. Clin. Psychol. 74, 870–879 (2006).
Gandhi, M., Yokoe, D. S. & Havlir, D. V. Asymptomatic transmission, the Achilles’ heel of current strategies to control Covid-19. N. Engl. J. Med. 382, 2158–2160 (2020).
Kimball, A. et al. Asymptomatic and presymptomatic SARS-CoV-2 infections in residents of a long-term care skilled nursing facility—King County, Washington, March 2020. MMWR Morb. Mortal. Wkly. Rep. 69, 377–381 (2020).
HHS details multiple COVID-19 testing statistics as national test. HHS (31 July 2020); https://www.hhs.gov/about/news/2020/07/31/hhs-details-multiple-covid-19-testing-statistics-as-national-test-volume-surges.html
CMS increases Medicare payment for high-production coronavirus lab tests. CMS (15 April 20); https://www.cms.gov/newsroom/press-releases/cms-increases-medicare-payment-high-production-coronavirus-lab-tests-0
Kliff, S. Most coronavirus tests cost about $100. Why did one cost $2,315? The New York Times (16 June 2020).
Pollitz, K. Free Coronavirus Testing for Privately Insured Patients? (Kasier Family Foundation, 2020); https://www.kff.org/coronavirus-policy-watch/free-coronavirus-testing-for-privately-insured-patients/
Larremore, D. B. et al. Test sensitivity is secondary to frequency and turnaround time for COVID-19 surveillance. Preprint at medRxiv https://doi.org/10.1101/2020.06.22.20136309 (2020).
Paltiel, A. D., David Paltiel, A., Zheng, A. & Walensky, R. P. Assessment of SARS-CoV-2 screening strategies to permit the safe reopening of college campuses in the United States. JAMA Netw. Open 3, e2016818 (2020).
Jones, E. M. et al. Octant SwabSeq Testing (2020); https://www.notion.so/Octant-SwabSeq-Testing-9eb80e793d7e46348038aa80a5a901fd
Jones, E. M. et al. A scalable, multiplexed assay for decoding GPCR-ligand interactions with RNA sequencing. Cell Syst. 8, 254–260 (2019).
Schmid-Burgk, J. L. et al. LAMP-Seq: population-scale COVID-19 diagnostics using a compressed barcode space. Preprint at bioRxiv https://doi.org/10.1101/2020.04.06.025635 (2020).
COVIDSeq Test (RUO and PEO Versions) (Illumina, 2020); https://www.illumina.com/products/by-type/clinical-research-products/covidseq.html
Bioinnovation’s DxSeqTM Sequences Filoviruses (Rapid Microbiology, 2014); https://www.rapidmicrobiology.com/news/bioinnovations-dxseq-sequences-filoviruses
Booeshaghi, A. S. et al. Reliable and accurate diagnostics from highly multiplexed sequencing assays. Sci. Rep. 10, 21759 (2020).
Relich, R. F. Preparation Of Viral Transport Medium (CDC, 2020); https://www.cdc.gov/coronavirus/2019-ncov/downloads/Viral-Transport-Medium.pdf
Information for Laboratories about Coronavirus (COVID-19) (Centers for Disease Control and Prevention, 2020); https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html
Ranoa, D. R. E. et al. Saliva-based molecular testing for SARS-CoV-2 that bypasses RNA extraction. Preprint at bioRxiv https://doi.org/10.1101/2020.06.18.159434 (2020).
COVID-19 Testing at Broad (Broad Institute, 2020); https://covid-19-test-info.broadinstitute.org/
iSWAB Rack Format (Mawi DNA Technologies, 2019); https://mawidna.com/our-products/iswab-rack-format/
TaqPath COVID-19 Multiplex Diagnostic Solution (Thermo Fisher Scientific, 2020).
Clark, T. W. et al. Diagnostic accuracy of the FebriDx host response point-of-care test in patients hospitalised with suspected COVID-19. J. Infect. https://doi.org/10.1016/j.jinf.2020.06.051 (2020).
Yujian, L. & Bo, L. A normalized Levenshtein distance metric. IEEE Trans. Pattern Anal. Mach. Intell. 29, 1091–1095 (2007).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Quail, M. A., Swerdlow, H. & Turner, D. J. Improved protocols for the illumina genome analyzer sequencing system. Curr. Protoc. Hum. Genet. 18, 62:18.2.1–18.2.27 (2009).
Morgan, M. et al. ShortRead: a bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608 (2009).
Van der Loo, M. P. J. The stringdist package for approximate string matching. R J. 6, 111–122 (2014).
Booeshaghi, A. S. et al. Reliable and accurate diagnostics from highly multiplexed sequencing assays. Sci. Rep. 10, 21759 (2020).
Melsted, P. et al. Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-00870-2 (2021).
Acknowledgements
We thank J. Semel for her support. We also thank the staff at the Held Foundation and the Carol Moss Foundation for their support of this project; staff at the UCLA David Geffen School of Medicine’s Dean’s Office for their support; Fast Grants Inc. for funding this work; L. Starita, B. Martin, J. Gehring, S. Srivatsan, J. Shendure and the members of the Covid Testing Scaleup Slack for their input, guidance and openness in sharing their processes; M. Berro for her guidance with the FDA EUA201963; the clinical laboratory scientists at the UCLA Clinical Microbiology laboratory for their assistance in collecting and processing the remnant specimens and data; our staff at the UCLA SwabSeq COVID19 Testing laboratory for deploying our CLIA test; and L. Yost and A. Martin for their advice and guidance during our scaling process. This work was supported by funding from the Howard Hughes Medical Institute (to L.K.) and DP5OD024579 (to V.A.A.). I.L. is supported by T32GM008042. Figures 1a,f and 3b created with BioRender.com.
Author information
Authors and Affiliations
Contributions
J.S.B. and V.A.A. wrote the manuscript with assistance from C.L., J.F., L.K., E.E., E.M.J., A.R.C., N.B.L., M.G. and S.Kosuri. E.M.J., A.R.C., N.B.L., M.G., S.W.S., J.S.B. and S.Kosuri designed barcodes and performed early testing and analysis of protocols and reagents. C.L., Y.Y., Y.Z., L.G., R.D. and M.J.B. provided early guidance and key automation resources. E.E., D.H., N.L. and C.K. developed the registration webapp and IT infrastructure. L.S., C.M., M.G., E.M.J., N.B.L., S.Kosuri, I.L., O.F.B., V.A.A. and J.S.B. performed and analysed experiments. A.S.B. and L.P. analysed misassignment of index barcodes. V.A.A., O.B.G., S.C., E.E.H., G.O. and B.J.C. collected and processed clinical samples. D.M. optimized operational protocols and D.M., S.Kay, M.L., T.L. and E.E. optimized scale up. E.E., L.K., J.F., C.L., Y.Y., Y.Z. and J.B. provided helpful insights into protocols, software, and development and optimization of our specimen collection and handling. F.Y., E.M.T., K.M.K., J.P. and M.H. developed the diversified S standard mixture, N1 primers and flu primers.
Corresponding authors
Ethics declarations
Competing interests
E.M.J., M.G., N.B.L., S.W.S., F.Y., E.M.T., K.M.K., J.P. and S.Kosuri are employed by and hold equity in Octant Inc., J.S.B. consults for and holds equity in Octant Inc., and A.R.C. holds equity in Octant Inc, which initially developed SwabSeq and has filed for patents for some of the work here, although they have been made available under the Open COVID License (https://www.notion.so/Octant-COVID-License-816b04b442674433a2a58bff2d8288df).
Additional information
Peer review information Nature Biomedical Engineering thanks Enzo Poirier and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary results, methods, figures, tables and references.
Supplementary Table 1
List of primers used.
Supplementary Table 2
List of the instrumentation and software used.
Supplementary Table 3
Precision and repeatability for purified and extraction-free saliva samples.
Supplementary Table 4
Tool to map scanned 96-well barcode racks to physical locations in 384-well plates.
Supplementary Table 5
Template to organize the construction of pilot SwabSeq experiments.
Rights and permissions
About this article

Cite this article
Bloom, J.S., Sathe, L., Munugala, C. et al. Massively scaled-up testing for SARS-CoV-2 RNA via next-generation sequencing of pooled and barcoded nasal and saliva samples. Nat Biomed Eng 5, 657–665 (2021). https://doi.org/10.1038/s41551-021-00754-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-021-00754-5
This article is cited by
-
RUBICON: a framework for designing efficient deep learning-based genomic basecallers
Genome Biology (2024)
-
Seeing Clearly in the Twilight: The Clinical and Forensic Relevance of the Indeterminate/Borderline Range in Multivariate Models of Performance Validity Assessment
Psychological Injury and Law (2024)
-
DeepSARS: simultaneous diagnostic detection and genomic surveillance of SARS-CoV-2
BMC Genomics (2022)
-
Microfluidic space coding for multiplexed nucleic acid detection via CRISPR-Cas12a and recombinase polymerase amplification
Nature Communications (2022)
-
Smart pooling: AI-powered COVID-19 informative group testing
Scientific Reports (2022)
